
Welcome To The Alzheimer’s Solution

Introducing BrainDefend®
A Personalized Body-Brain Health and
Renewal Program designed to protect
and optimize your cognitive health as you age.
THE ALZHEIMER'S SOLUTION
Latest News from the Blog
Late-Onset Alzheimer’s Disease—The Hidden Midlife Crisis
Midlife may be a critical juncture in one's life and a time for contemplating what the rest of it will look like. It may be a time of reflection on time well spent or sheer regret for not having done enough of what you really love to do. Midlife may also be a turning...
Polyphenols in the Protection Against Cardiovascular Disease and Dementia
In a recently published study—NutriNet-Santé (10/2018)—that included the analysis of dietary records of 84,158 French adults that spanned between May 2009 and June 20017— the study authors concluded found that “Higher intakes of polyphenols, especially anthocyanins...
Inflammation and Alzheimer’s Disease—Cause, or Effect?
The role of chronic inflammation in degenerative diseases associated with aging is considered to be a primary vector for the progression of neurodegenerative disorders and a powerful factor that underlies their etiology. One needs only to look at the leading causes of...
THE ALZHEIMER'S SOLUTION
Save Your Brain - Save Your Memories - Save Your Life
The Alzheimer’s Solution Revolution Podcast
THE ALZHEIMER'S SOLUTION
Think Ahead for a Better Brain Tomorrow!
MEMBER ACCESS: PFAS—The Little-Known Forever Chemicals are Emerging as Dangerous Neurotoxicants

MEMBER ACCESS: Mercury Depletes Glutathione Peroxidase-Toxic Mechanisms In Alzheimer’s Disease Risk
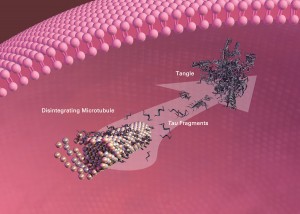
MEMBER ACCESS: Oxidative Stress and the Thromboxane Receptor—A Central Pivot in the Production of Neurofibrillary Tangles

MEMBER ACCESS: TREM2 genetic variants in Alzheimer’s disease

MEMBER ACCESS: Blueberry Polyphenols Protect the Brain from the Degenerative Processes Associated with Brain Aging and Alzheimer’s Disease

MEMBER ACCESS: Brain-Derived Neurotrophic Factor—Growth Factors in Neurogenesis and the Protection Against Alzheimer’s Disease Progression
Join Us & Get MEMBER Access – it’s FREE!

#HolidayBlessings #PeaceLoveProsperity
Wishing all of you a blessed day and a New Year filled with abundance❣️

0 CommentsComment on Facebook
#thankful #blessed
Wherever you are and however you celebrate Thanksgiving day, I hope the day brings you and your loved ones joy!
God bless all of you❣️
0 CommentsComment on Facebook
#alzheimersrisk #women #omega3
The article linked to here is a report on the association between deficits of dietary omega-3 fatty acids (O3s) and risk for Alzheimer's disease (AD) in women.
While that finding concerning a lower O3 in a woman's risk for AD, the same outcome was not evident in men.
The report on the study also cited the association between higher levels of saturated fatty acids (saturated fats/lipids) to omega-3s (unsaturated lipids ) as a feature of the lipid profiles in the women included in the study—no surprise there.
The report also included commentary from the senior author of the study, Cristina Legido-Quigley, PhD, who opined that "further work is needed ... Understanding how the disease works differently in women could help doctors tailor future treatments and health advice”.
True, but here is a significant factor not included in the study or report with regard to why omega-3s play a critical role in women's brains as they age.
First, omega-3s and other fats, including phospholipids (phosphatidylcholine), and cholesterol are critical constituents of brain structures such as the all-important cellular membranes that envelope neurons, and the fatty myelin insulation sheath that covers the long neuronal extensions—axons.
The myelin insulation sheath on axons is a vital structure that insulates and enhances the conduction of electrical signals in neuron-to-neuron communication and the vast networks that they comprise.
In women, estrogen governs glucose metabolism and also functions as a neuroprotective hormone.
As I described in my book, "The Diabetic Brain in Alzheimer's Disease", estrogen deficiency in post-menopausal women is a risk factor for deficits in glucose and mitochondrial energy metabolism (glucose hypometabolism).
To compensate for the glucose hypometabolism in the aging female brain, fats serve as an alternate fuel for energy metabolism, and studies have shown that myelin is cannibalized in order to supply fats for mitochondrial energy metabolism.
Thus, in post-menopausal and estrogen-deficient women, a failure in disease-modifying dietary and lifestyle interventions, or the recognition that hormone replacement therapy may be a suitable intervention, may result in a very vulnerable brain.
The destruction of myelin in neurodegenerative disorders is a neuropathological feature that characterizes multiple sclerosis and is a known component of the pathology associated with Alzheimer’s disease.
However, in the case of women and their risk for late-onset Alzheimer's disease (LOAD), ketogenic diet therapy and/or personalized dietary and supplemental fats (omega-3s, phospholipids) may function as the recipe for myelin preservation and Alzheimer’s disease prevention.
Lastly, O3 supplementation is not the same, and the role of fat transport in the body and brain is an important component to the effective delivery of fats such as O3s to the tissues that depend on them.
How well O3s such as EPA and DHA are delivered to the brain is, in large part, dependent on the form of O3.
For example, O3s bound to triglycerides are not transported across the blood-brain barrier (BBB), as well as other forms, such as O3 that is bound to phospholipids (lysophosphatidylcholine).
The uptake of DHA or EPA bound to lysophosphatidylcholine across the BBB is facilitated via a specialized transporter (MFSD2A).
To get a more comprehensive audio overview on the role of glucose hypometabolism in a woman's risk for LOAD, please listen to my podcast titled:
"Glucose Hypometabolism, Impairments in Mitochondrial Function, and Oxidative Stress as Major Risk Factors for Alzheimer’s Disease" @ podcasts.apple.com/us/podcast/glucose-hypometabolism-impairments-in-mitochondrial/id1614865183
Omega-3 Fatty Acids and AD Risk: What Do the Data Show?
www.medscape.com
A clear deficit in highly unsaturated lipids — including omega-3 fatty acids — found in women with AD but not men, could open new paths for research and possibly prevention.0 CommentsComment on Facebook
#cholesteroldrugtrial #Alzheimersrisk
There is an immense body of evidence that links the progression of atherosclerosis and cardiovascular disease in aging individuals with an increased risk for vascular dementia and late-onset Alzheimer's disease (LOAD).
In my book, "The Diabetic Brain in Alzheimer's Disease", I detail the role of type 2 diabetes and cardiovascular disease (cardiometabolic disease) in the development of vascular dementia and LOAD later in life.
Ditto for the role of the ApoE4 genetic variant in the risk for cardiometabolic disease, vascular dementia, and LOAD, which brings us to yet another investigation drug trial that explores the possible use of a cholesterol (LDL) lowering drug in the treatment of LOAD.
For a more comprehensive review of the role of ApoE4, and other genes and genetic variants associated with cholesterol metabolism and transport, please listen in to my podcast on the topic at:
podcasts.apple.com/us/podcast/genetic-variants-in-the-risk-for-late-onset/id1614865183?i=10005599...
BTW, such studies that explored the use of statins in the treatment for Alzheimer's disease have at best shown mixed outcomes—with studies indicating that statin users have a reduced risk for dementia and LOAD, and other studies unequivocally indicating no dementia or Alzheimer's prevention outcomes.
Such mixed research findings is not surprising in light of fact that LOAD is a multifactorial disease, and cholesterol dysmetabolism is just one constituent of a very complex disorder.
So with that backstory in mind, here is yet another cholesterol drug report with regard to the treatment of Alzheimer's disease.
The drug—obicetrapib, which is in a class of cholesteryl ester transfer protein (CETP) inhibitors, targets cholesterol transfer metabolism—HDL to LDL transfer.
CTEP inhibitors are often lumped into the class of lipid-lowering drugs.
Regardless, CETP inhibitors benefits cholesterol levels by raising HDL and lowering LDL cholesterol, albeit previous generations (e.g., dalcetrapib, torcetrapib) of CTEP inhibitor trials were halted due to a lack of efficacy and an increased risk of mortality.
In a report linked to here regarding "new research" obicetrapib "significantly slowed Alzheimer’s disease (AD)".
An important finding in the study found a " 20% improvement in levels" of an AD biomarker—tau protein (p-tau217) in carriers of the ApoE4 genetic risk variant that agian, is strongly associated with an increased risk for LOAD.
Note, the commentary in the report regarding the lack of sufficient cholesterol lowering statin therapy efficacy in a previous study (BROADWAY), and the added benefit of obicetrapib to the treatment protocol.
Nevertheless, in the obicetrapib trial group there was a significant impact on the lowering LOAD risk biomarkers such as p-tau217 to the beta-amyloid (Aβ) 42/40 ratio biomarker.
Impressive findings but there is much more to the role of of cholesterol dysmetabolism and the ApoE4 risk variant in the risk for vascular dementia and LOAD that warrants retelling here.
First, the obicetrapib trial participants were 66-70 years of age.
I've previously posted on how important it is to begin tracking your risk for dementia and LOAD earlier in life which empowers a prevention-minded approach.
LOAD and cardiometabolic disease has at lease a 20 to 30 year trajectory before a diagnosis of a more significant disease progression. and the complications that go along with both.
In my book, I reaffirm the critical strategy of biomarker assessment and tracking for individuals with an increased risk for dementia and LOAD due to age-related diseases such as cardiometabolic disease.
I also emphasize that midlife is a critical juncture in the trajectory of age-related disease and the risk for dementia and LOAD—if you wait 'till you are 66-70 years old, you've likely missed a great deal of disease prevention opportunities as cardiometabolic disease and LOAD begins much earlier in life.
Think ahead. If there is a family history of cardiometabolic disease, dementia or AD, then take control of your diet and lifestyle risk factors which leverages a prevention lifestyle.
And, press you physician to track your LOAD and cardiometabolic risk biomarkers!
BrainDefend®
Experimental Med May Slow Alzheimer’s Biomarker Progression
www.medscape.com
The experimental cholesterol drug obicetrapib slowed Alzheimer’s biomarkers, over 12 months in patients with cardiovascular disease, offering a potential new therapy.0 CommentsComment on Facebook
THE ALZHEIMER'S SOLUTION
TestimonialsWhat a tremendous resource! Thank you for putting all of this great research in one place! I am also extremely interested in warding off a family history of Alzheimer’s and Dementia. Medicine can be so very powerful when we are able to identify the biochemical inefficiencies, apply specific dietary and nutritional remedies and compile a protocol to heal not just an individual, but generations. Thank you Ralph!
This is an incredible article! Very powerful information! Thank you so much for putting it together the way that you have. This information needs to get out to the masses. You explained that very well.
Absolutely the BEST article I have read on the insulin/AD connection. As always, your brilliance is very much appreciated!
Thank you so much for this well written article. Looking forward to future updates. Thanks for this well illustrated, researched, and detailed yet succinct article.